Protein phosphorylation is one of the most critical post-translational modifications to regulate cellular function. Measuring the phosphorylation event at the proteome scale provides a system view for activated signaling pathway. A pioneering study by an international research group which is led by Dr. Yu-Ju Chen from the Institute of Chemistry, Academia Sinica and Prof Yasushi Ishihama from the Graduate School of Pharmaceutical Sciences, Kyoto University has developed a novel mass spectrometry-based technique for the first measurement on the basal level of phosphorylation stoichiometry in a single human phosphoproteome and identified potential molecular changes associated with gefitinib resistance in lung cancer cells. The study was published online on March 27, 2015 in the journal Nature Communications.
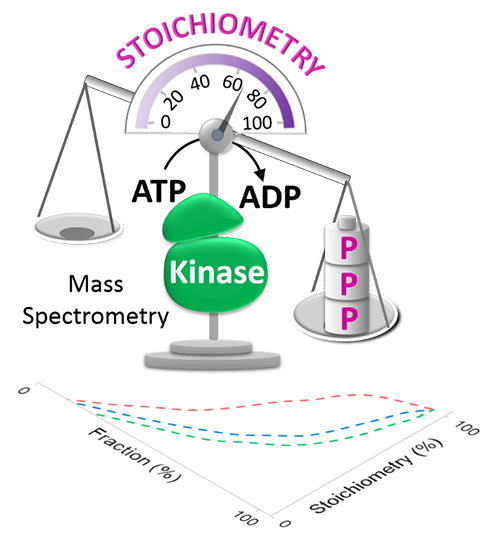
Deregulated signaling through protein phosphorylation is intimately linked to pathogenesis of human disease and is one of the most clinically accessed post-translational modifications for developing new therapeutic strategies. The signal-induced alteration in phosphoprotein is regulated either by upstream kinase/phosphatase activity to cause change in phosphorylation stoichiometry, defined as the ratio of the total amount of protein phosphorylated on a specific site to the total amount of protein, or by transcriptional regulation to modify protein abundance. However, traditional quantitative approach can only measure relative quantitation of phosphorylation events without information about the absolute stoichiometry of modification within proteins; direct measurement of phosphorylation stoichiometry, which allows digitizing the cellular signaling network, still remains a challenge by existing approaches.
To overcome the current bottleneck in accessing the stoichiometry of single-state human phosphoproteomes, we have developed a motif-targeting quantitative proteomic approach by integrating enzymatic kinase reaction and isotope-based quantitative proteomic strategy. The quantitation accuracy and sensitivity of this approach was demonstrated on the proof-of-concept experiments in lung cancer cell; phosphorylation stoichiometry of >1000 phosphorylation sites including 366 low abundant tyrosine phosphorylation sites were successfully measured with high reproducibility. To our knowledge, this approach reveals the first large-scale measurement on the basal level of phosphorylation stoichiometry in a single state human phosphoproteome.
This research group further applied this developed motif targeting quantitative approach for phosphorylation stoichiometry profiling of drug resistance/sensitive lung cancer cells. On the comparison of TKI (tyrosine kinase inhibitor) sensitive (PC9) and resistance lung cancer cell (PC9/gef.), the quantitative information not only illuminated that the post-translational phosphorylation changes are significantly more dramatic than those at protein as well as mRNA levels, but also suggested potential drug-targeting proteins in the kinase-substrate network associated with acquired drug-resistance. We expected that this newly developed approach will have wide applications to provide system-wide maps of protein phosphorylation stoichiometry for either single or multiple cellular states under physiological or pathological regulation.
The complete article is available at the Nature Communications journal website at:
http://www.nature.com/ncomms/index.html.
The complete list of authors is: Chia-Feng Tsai, Yi-Ting Wang, Hsin-Yung Yen, Chih-Chiang Tsou, Wei-Chi Ku, Pei-Yi Lin, Hsuan-Yu Chen, Alexey I. Nesvizhskii, Yasushi Ishihama and Yu-Ju Chen
Media contacts:
Dr. Yu-Ju Chen, Research Fellow, Institute of Chemistry, Academia Sinica, yujuchen@gate.sinica.edu.tw (Tel) +886-2-2789-8660